
MaldiAMRKit - Exploration & Similarity#
This notebook demonstrates how to use MaldiAMRKit’s exploratory visualisation and spectral similarity functions to inspect MALDI-TOF datasets before building classifiers.
It covers:
PCA scatter plots coloured by metadata
t-SNE embeddings
UMAP embeddings (requires
pip install maldiamrkit[batch])Spectral similarity: pairwise distance matrices, heatmaps, clustering, and dendrograms
Brief introduction to batch effect correction with
combatlearn
Dataset#
This notebook uses the full multi-site version of the MALDI-Kleb-AI dataset (Rocchi et al., 2026; Zenodo DOI 10.5281/zenodo.17405072) - all three Italian centres (Rome, Milan, Catania), about 770 spectra. The other notebooks restrict themselves to the Rome sub-cohort, but exploration is precisely where you want the full multi-centre cohort: unsupervised methods will quickly tell you whether site / batch effects dominate the resistance signal.
The helper in `notebooks/_demo.py <_demo.py>`__ caches the 370 MB tarball under ~/.cache/maldiamrkit/ on first use.
[1]:
import pathlib
import sys
from maldiamrkit.visualization import plot_pca, plot_tsne
sys.path.insert(0, str(pathlib.Path.cwd())) # _demo.py sits next to this notebook
from _demo import load_maldi_kleb_ai
/home/ettore/.venvs/maldiamrkit/lib/python3.10/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Load the Dataset#
Exploration is the one notebook where the full multi-site cohort is more interesting than the Rome-only slice: with three acquisition centres (Rome, Milan, Catania) we can see whether unsupervised structure tracks the resistance phenotype or the acquisition site. Pass city=None to load all three centres.
[2]:
ds = load_maldi_kleb_ai(antibiotic="Amikacin", city=None, verbose=True)
data = ds.maldi_set
labels = ds.meta["Amikacin"]
sites = ds.meta["City"]
print(f"Samples: {ds.X.shape[0]}, Features: {ds.X.shape[1]}")
print(f"Labels: {labels.value_counts().to_dict()}")
print(f"Sites: {sites.value_counts().to_dict()}")
Processing spectra: 100%|██████████| 743/743 [00:00<00:00, 4637.49spectrum/s]
Samples: 741, Features: 6000
Labels: {'S': 372, 'R': 369}
Sites: {'Rome': 470, 'Milan': 184, 'Catania': 87}
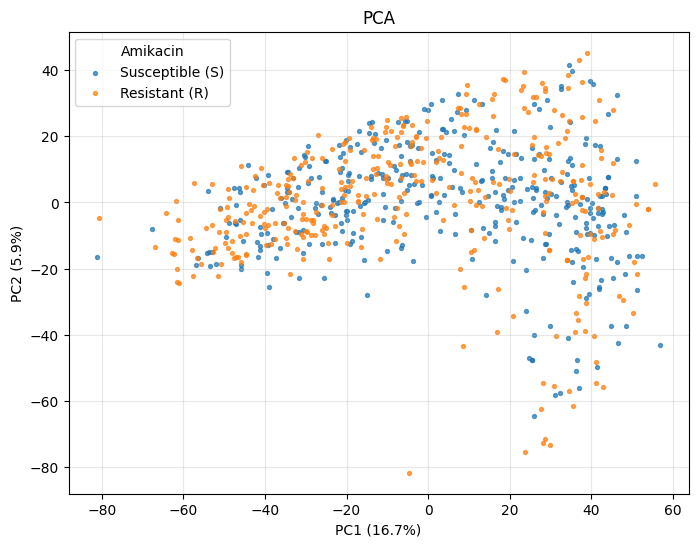
PCA#
PCA is the simplest starting point. Axis labels automatically show the explained variance percentage.
[3]:
_ = plot_pca(ds.X, color_by=labels)
You can pass any matplotlib keyword arguments through **pca_kwargs, or provide a custom palette:
[4]:
_ = plot_pca(
ds.X,
color_by=labels,
palette={"S": "#2196F3", "R": "#F44336"},
title="PCA - Amikacin Resistance (multi-site)",
alpha=0.8,
s=35,
)
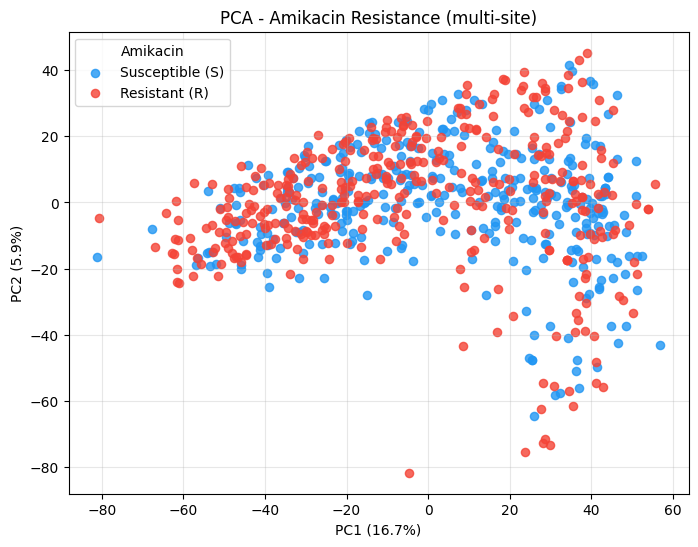
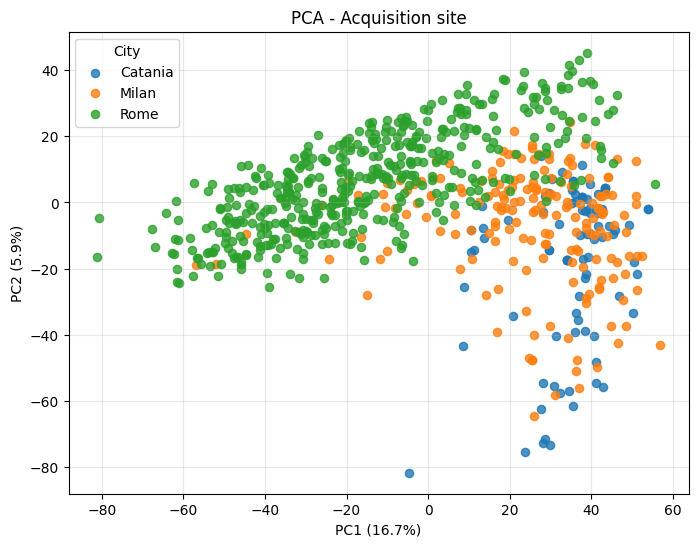
Now colour by the acquisition centre instead - if the three sites separate, you are seeing batch / instrument effects rather than biology, and the unsupervised plots are not telling you about resistance.
[5]:
_ = plot_pca(
ds.X,
color_by=sites,
title="PCA - Acquisition site",
alpha=0.8,
s=35,
)
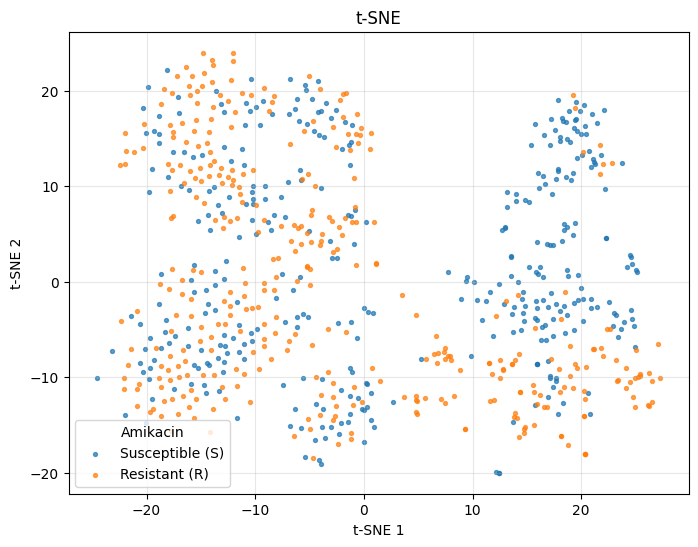
t-SNE#
For a non-linear embedding, use plot_tsne. With ~470 samples, perplexity in the 20-40 range is a reasonable default.
[6]:
_ = plot_tsne(ds.X, color_by=labels, perplexity=30, random_state=42)
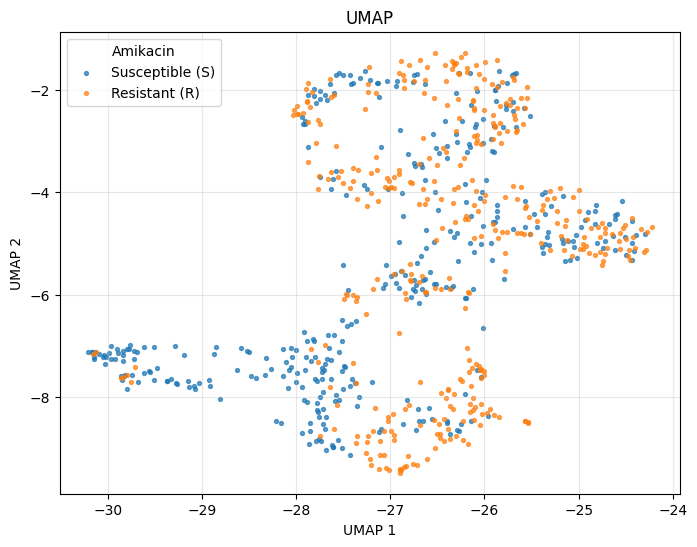
UMAP#
UMAP requires the optional umap-learn package:
pip install maldiamrkit[batch]
[7]:
try:
from maldiamrkit.visualization import plot_umap
_ = plot_umap(ds.X, color_by=labels, n_neighbors=15, random_state=42)
except ImportError as e:
print(e)
/home/ettore/.venvs/maldiamrkit/lib/python3.10/site-packages/umap/umap_.py:1952: UserWarning: n_jobs value 1 overridden to 1 by setting random_state. Use no seed for parallelism.
warn(
Spectral Similarity#
MaldiAMRKit provides spectral distance metrics, pairwise distance matrix computation, clustering algorithms, and dedicated plots for similarity analysis.
Available metrics (see SpectralMetric):
cosine - cosine distance (binned spectra)
spectral_contrast_angle - spectral contrast angle (binned spectra)
pearson - 1 - Pearson correlation (binned spectra)
wasserstein - Wasserstein / earth mover’s distance (raw spectra)
dtw - dynamic time warping distance (raw spectra)
For the pairwise heatmap and dendrogram we subsample to keep the figures readable; the same calls scale to the full 470-spectrum matrix.
[8]:
from maldiamrkit.similarity import (
cluster_metadata_concordance,
cluster_spectra,
hierarchical_clustering,
pairwise_distances,
plot_dendrogram,
plot_distance_heatmap,
silhouette_scores,
)
Pairwise Distance Matrix#
Compute a symmetric distance matrix from the binned feature matrix. For binned spectra, cosine distance is a natural choice.
[9]:
import numpy as np
rng = np.random.default_rng(0)
subset_idx = rng.choice(ds.X.shape[0], size=80, replace=False)
X_subset = ds.X.iloc[subset_idx]
labels_subset = labels.iloc[subset_idx]
D = pairwise_distances(X_subset, metric="cosine")
print(f"Distance matrix shape: {D.shape}")
print(f"Range: [{D[D > 0].min():.4f}, {D.max():.4f}]")
Distance matrix shape: (80, 80)
Range: [0.0449, 0.8068]
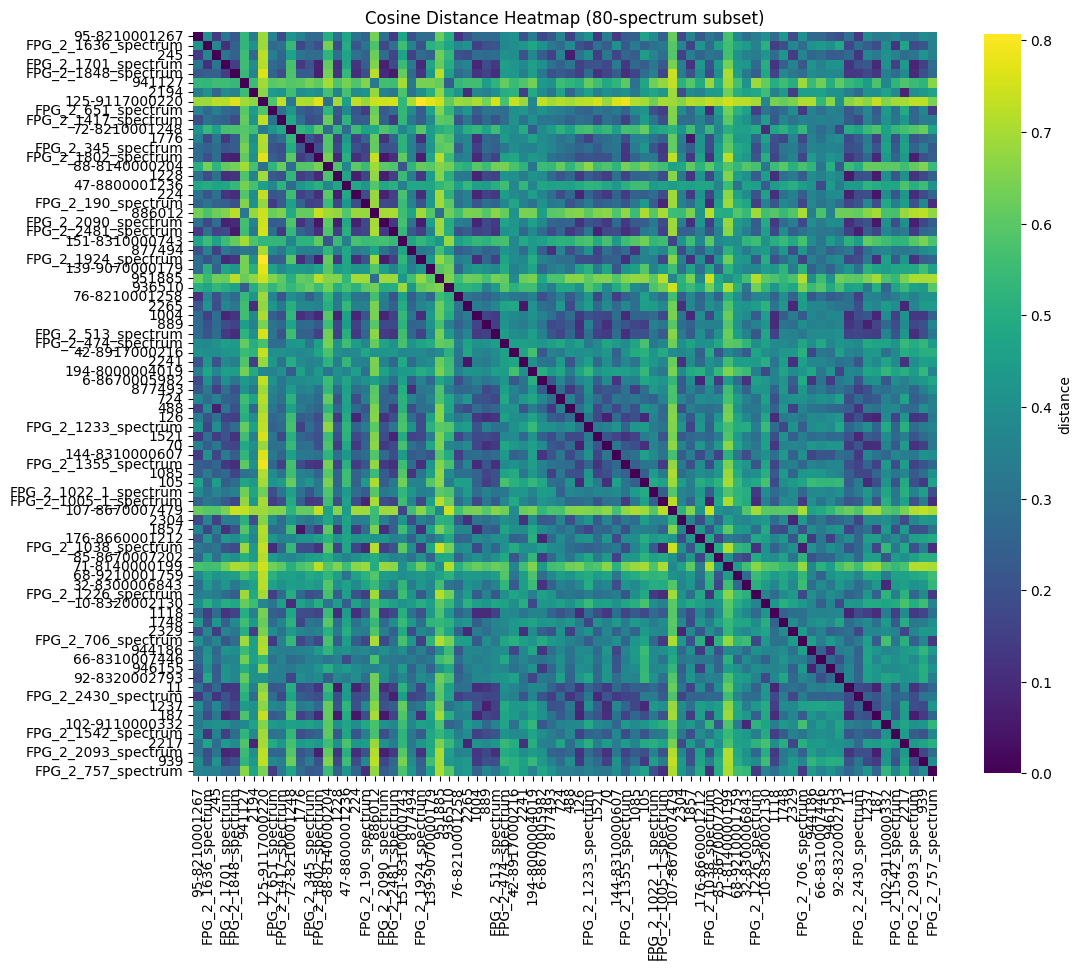
Distance Heatmap#
Visualise the pairwise distance matrix. Darker values indicate more similar spectra.
[10]:
_ = plot_distance_heatmap(
D,
labels=X_subset.index.tolist(),
title="Cosine Distance Heatmap (80-spectrum subset)",
)
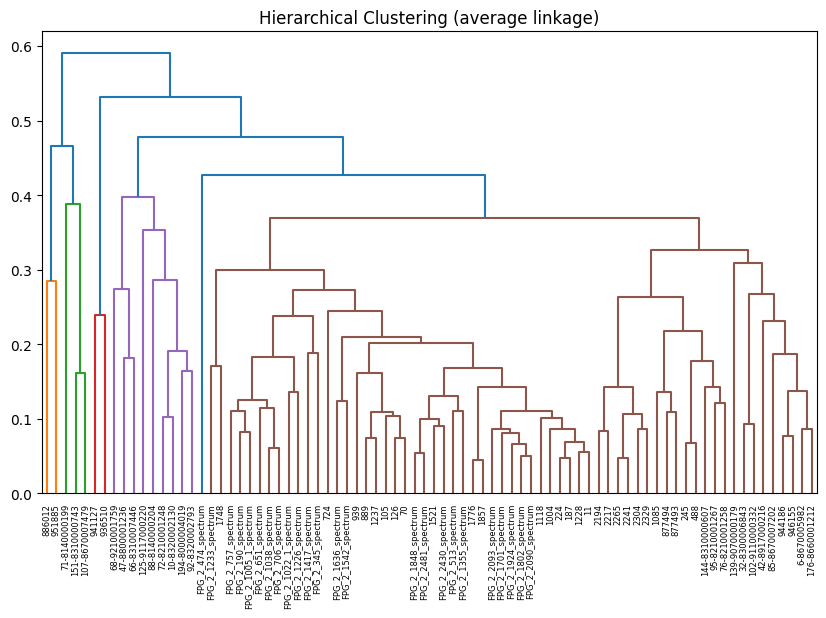
Hierarchical Clustering & Dendrogram#
Build a linkage matrix and visualise the cluster hierarchy.
[11]:
linkage = hierarchical_clustering(D, method="average")
_ = plot_dendrogram(
linkage,
labels=X_subset.index.tolist(),
title="Hierarchical Clustering (average linkage)",
)
Cluster Assignment & Evaluation#
Use cluster_spectra for a one-call clustering workflow. We ask for three clusters - matching the three acquisition centres - and then score the partition against both the resistance phenotype and the site label with silhouette scores and metadata concordance (ARI, NMI). This is a clean diagnostic: if clusters track the site but not the phenotype, batch effects are dominating the unsupervised signal - a classic motivator for ComBat-style correction.
We use method='kmedoids' here: with very uneven cluster sizes (Rome alone is ~470 spectra) average-linkage hierarchical clustering tends to peel off a couple of singletons instead of recovering the three centres. K-medoids respects the requested cluster count and produces balanced groups that closely match the site labels.
[12]:
import pandas as pd
D_full = pairwise_distances(ds.X, metric="cosine")
cluster_labels = cluster_spectra(
D_full,
method="kmedoids",
n_clusters=3,
random_state=42,
)
sil = silhouette_scores(D_full, cluster_labels)
print(f"Silhouette score: {sil:.3f}")
conc_pheno = cluster_metadata_concordance(cluster_labels, labels)
print("\nConcordance vs. Amikacin (R/S):")
print(f" Adjusted Rand Index: {conc_pheno['adjusted_rand_index']:.3f}")
print(f" Normalized Mutual Info: {conc_pheno['normalized_mutual_info']:.3f}")
conc_site = cluster_metadata_concordance(cluster_labels, sites)
print("\nConcordance vs. acquisition site (City):")
print(f" Adjusted Rand Index: {conc_site['adjusted_rand_index']:.3f}")
print(f" Normalized Mutual Info: {conc_site['normalized_mutual_info']:.3f}")
print("\nClusters x City:")
pd.crosstab(
pd.Series(cluster_labels, index=sites.index, name="Cluster"),
sites,
)
Silhouette score: 0.294
Concordance vs. Amikacin (R/S):
Adjusted Rand Index: 0.029
Normalized Mutual Info: 0.042
Concordance vs. acquisition site (City):
Adjusted Rand Index: 0.688
Normalized Mutual Info: 0.541
Clusters x City:
[12]:
| City | Catania | Milan | Rome |
|---|---|---|---|
| Cluster | |||
| 0 | 7 | 7 | 439 |
| 1 | 80 | 110 | 21 |
| 2 | 0 | 67 | 10 |
Comparing Metrics#
Try different distance metrics to see which one recovers the site structure most cleanly. We compare each metric against both the resistance phenotype and the acquisition site.
[13]:
results = []
for metric in ["cosine", "spectral_contrast_angle", "pearson"]:
D_m = pairwise_distances(ds.X, metric=metric)
cl = cluster_spectra(D_m, method="kmedoids", n_clusters=3, random_state=42)
sil_m = silhouette_scores(D_m, cl)
conc_pheno = cluster_metadata_concordance(cl, labels)
conc_site = cluster_metadata_concordance(cl, sites)
results.append(
{
"metric": metric,
"silhouette": sil_m,
"ARI_pheno": conc_pheno["adjusted_rand_index"],
"NMI_pheno": conc_pheno["normalized_mutual_info"],
"ARI_site": conc_site["adjusted_rand_index"],
"NMI_site": conc_site["normalized_mutual_info"],
}
)
pd.DataFrame(results).set_index("metric")
[13]:
| silhouette | ARI_pheno | NMI_pheno | ARI_site | NMI_site | |
|---|---|---|---|---|---|
| metric | |||||
| cosine | 0.294047 | 0.029394 | 0.041665 | 0.687924 | 0.541444 |
| spectral_contrast_angle | 0.152448 | 0.019211 | 0.016085 | 0.449982 | 0.410990 |
| pearson | 0.250229 | 0.016489 | 0.013949 | 0.391881 | 0.361815 |
Batch Effect Correction#
When combining data from multiple sites or instruments, batch effects can dominate the signal. MaldiAMRKit on its own focuses on the single- site case (that is why this notebook restricts to Rome). For multi- centre harmonisation use combatlearn directly, or use the dedicated MaldiBatchKit sister package which builds on top of it:
from combatlearn import Combat
combat = Combat(method='fortin')
combat.fit(X, y=batch_labels)
X_corrected = combat.transform(X, y=batch_labels)
Install with pip install maldiamrkit[batch]. See the combatlearn documentation for full usage.
Rocchi, E., Nicitra, E., Calvo, M. et al. Combining mass spectrometry and machine learning models for predicting Klebsiella pneumoniae antimicrobial resistance: a multicenter experience from clinical isolates in Italy. BMC Microbiol (2026). doi:10.1186/s12866-025-04657-2