
MaldiAMRKit - Differential Analysis#
This notebook demonstrates the maldiamrkit.differential module for identifying m/z bins that differ significantly between resistant (R) and susceptible (S) groups, and for comparing significant peaks across multiple drugs.
It covers:
DifferentialAnalysison a binned feature matrix (Mann-Whitney U or Welch’s t-test)Multiple-testing correction (FDR-BH, FDR-BY, Bonferroni), log2 fold change, Cohen’s d
Ranking peaks with
top_peaks()and filtering withsignificant_peaks()AMR-aware plots:
plot_volcano,plot_manhattanMulti-drug comparison via
compare_drugs+plot_drug_comparison(heatmap / UpSet)
Dataset#
These notebooks use the MALDI-Kleb-AI dataset (Rocchi et al., 2026; Zenodo DOI 10.5281/zenodo.17405072), a curated archive of MALDI-TOF spectra of Klebsiella clinical isolates from three Italian centres (Rome, Milan, Catania) with Amikacin / Meropenem resistance annotations. For simplicity we restrict the demo to the Rome sub-cohort (single site, no batch correction needed). The helper in `notebooks/_demo.py <_demo.py>`__ caches the 370
MB tarball under ~/.cache/maldiamrkit/ (or $MALDIAMRKIT_CACHE_DIR) on first use.
Load the Dataset#
We load the Rome cohort and use LabelEncoder(intermediate='drop') to build a binary label vector. The Zenodo dataset also ships a Meropenem annotation, which we will use later for the multi-drug comparison.
[1]:
import pathlib
import sys
import pandas as pd
from maldiamrkit.susceptibility import LabelEncoder
sys.path.insert(0, str(pathlib.Path.cwd())) # _demo.py sits next to this notebook
from _demo import load_maldi_kleb_ai
ds = load_maldi_kleb_ai(antibiotic="Amikacin", verbose=True)
encoder = LabelEncoder(intermediate="nan") # map 'I' to NaN so lengths stay aligned
y = (
pd.Series(
encoder.fit_transform(ds.meta["Amikacin"]),
index=ds.meta.index,
name="Amikacin",
)
.dropna()
.astype(int)
)
X = ds.X.loc[y.index]
print(f"X shape: {X.shape}")
print(f"Class counts: {y.value_counts().to_dict()}")
/home/ettore/.venvs/maldiamrkit/lib/python3.10/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Processing spectra: 0%| | 0/472 [00:00<?, ?spectrum/s]
Processing spectra: 98%|█████████▊| 464/472 [00:00<00:00, 4632.35spectrum/s]
Processing spectra: 100%|██████████| 472/472 [00:00<00:00, 4600.71spectrum/s]
X shape: (470, 6000)
Class counts: {1: 273, 0: 197}
Run the Differential Analysis#
Instantiate DifferentialAnalysis with X and y and call .run(). The default test is Mann-Whitney U (non-parametric, recommended for MALDI peak intensities) with Benjamini-Hochberg FDR correction.
[2]:
from maldiamrkit.differential import DifferentialAnalysis
analysis = DifferentialAnalysis(X, y).run(
test="mann_whitney",
correction="fdr_bh",
)
analysis.results.sort_values("adjusted_p_value", ascending=True).head()
[2]:
| mz_bin | mean_r | mean_s | fold_change | p_value | adjusted_p_value | effect_size | |
|---|---|---|---|---|---|---|---|
| 904 | 4712.0 | 0.000492 | 0.000161 | 1.611970 | 1.031418e-07 | 0.000155 | 0.472830 |
| 905 | 4715.0 | 0.000506 | 0.000162 | 1.642962 | 3.811623e-08 | 0.000155 | 0.546662 |
| 901 | 4703.0 | 0.000284 | 0.000120 | 1.241332 | 7.021650e-08 | 0.000155 | 0.522023 |
| 3038 | 11114.0 | 0.000125 | 0.000080 | 0.652812 | 8.531360e-08 | 0.000155 | 0.589519 |
| 3037 | 11111.0 | 0.000102 | 0.000067 | 0.604420 | 1.692839e-07 | 0.000197 | 0.545698 |
The results table has one row per m/z bin with:
mz_bin- the bin label (m/z value as stored inX.columns)mean_r,mean_s- mean intensity in each groupfold_change-log2((mean_r + eps) / (mean_s + eps))p_value- raw test p-valueadjusted_p_value- p-value after multiple-testing correctioneffect_size- Cohen’s d between the two groups
Construct directly from a MaldiSet#
When the antibiotic column already stores numeric binary labels (0 / 1), you can skip the manual X / y extraction:
analysis = DifferentialAnalysis.from_maldi_set(
ds.maldi_set, antibiotic='Amikacin'
).run()
In our case the labels are the strings 'R' / 'S', so we keep the explicit LabelEncoder step above.
[3]:
print(f"{len(analysis.results)} bins tested")
print(f"Smallest adjusted p-value: {analysis.results['adjusted_p_value'].min():.3e}")
print(f"#adj <= 0.05: {(analysis.results['adjusted_p_value'] <= 0.05).sum()}")
6000 bins tested
Smallest adjusted p-value: 1.547e-04
#adj <= 0.05: 22
Top Peaks and Significance Filtering#
[4]:
analysis.top_peaks(n=10)
[4]:
| mz_bin | mean_r | mean_s | fold_change | p_value | adjusted_p_value | effect_size | |
|---|---|---|---|---|---|---|---|
| 901 | 4703.0 | 0.000284 | 0.000120 | 1.241332 | 7.021650e-08 | 0.000155 | 0.522023 |
| 904 | 4712.0 | 0.000492 | 0.000161 | 1.611970 | 1.031418e-07 | 0.000155 | 0.472830 |
| 905 | 4715.0 | 0.000506 | 0.000162 | 1.642962 | 3.811623e-08 | 0.000155 | 0.546662 |
| 3038 | 11114.0 | 0.000125 | 0.000080 | 0.652812 | 8.531360e-08 | 0.000155 | 0.589519 |
| 3036 | 11108.0 | 0.000123 | 0.000083 | 0.562401 | 2.293709e-07 | 0.000197 | 0.518235 |
| 3037 | 11111.0 | 0.000102 | 0.000067 | 0.604420 | 1.692839e-07 | 0.000197 | 0.545698 |
| 3039 | 11117.0 | 0.000127 | 0.000083 | 0.604224 | 2.099257e-07 | 0.000197 | 0.558619 |
| 902 | 4706.0 | 0.000376 | 0.000140 | 1.430031 | 4.248146e-07 | 0.000319 | 0.491018 |
| 900 | 4700.0 | 0.000243 | 0.000120 | 1.017319 | 7.629639e-07 | 0.000509 | 0.442172 |
| 903 | 4709.0 | 0.000417 | 0.000150 | 1.470420 | 2.874043e-06 | 0.001724 | 0.464538 |
[5]:
sig = analysis.significant_peaks(fc_threshold=1.0, p_threshold=0.05)
print(f"{len(sig)} peaks survive |log2FC| >= 1 and adj.p <= 0.05")
sig.head(10)
8 peaks survive |log2FC| >= 1 and adj.p <= 0.05
[5]:
| mz_bin | mean_r | mean_s | fold_change | p_value | adjusted_p_value | effect_size | |
|---|---|---|---|---|---|---|---|
| 0 | 4526.0 | 0.000472 | 0.000097 | 2.282285 | 4.044768e-05 | 0.014276 | 0.394177 |
| 1 | 4700.0 | 0.000243 | 0.000120 | 1.017319 | 7.629639e-07 | 0.000509 | 0.442172 |
| 2 | 4703.0 | 0.000284 | 0.000120 | 1.241332 | 7.021650e-08 | 0.000155 | 0.522023 |
| 3 | 4706.0 | 0.000376 | 0.000140 | 1.430031 | 4.248146e-07 | 0.000319 | 0.491018 |
| 4 | 4709.0 | 0.000417 | 0.000150 | 1.470420 | 2.874043e-06 | 0.001724 | 0.464538 |
| 5 | 4712.0 | 0.000492 | 0.000161 | 1.611970 | 1.031418e-07 | 0.000155 | 0.472830 |
| 6 | 4715.0 | 0.000506 | 0.000162 | 1.642962 | 3.811623e-08 | 0.000155 | 0.546662 |
| 7 | 4718.0 | 0.000366 | 0.000140 | 1.388156 | 1.361776e-05 | 0.006809 | 0.524848 |
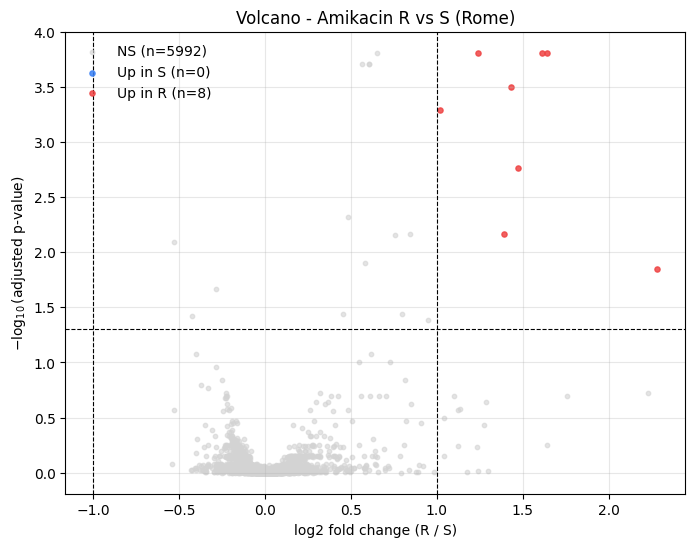
Volcano Plot#
plot_volcano shows log2 fold change on the x axis and -log10(adjusted p-value) on the y axis. Points are coloured by direction:
red: up in resistant (
fold_change > fc_thresholdandadj.p <= p_threshold)blue: up in susceptible (
fold_change < -fc_thresholdandadj.p <= p_threshold)grey: non-significant
[6]:
from maldiamrkit.differential import plot_volcano
_ = plot_volcano(
analysis.results,
fc_threshold=1.0,
p_threshold=0.05,
title="Volcano - Amikacin R vs S (Rome)",
)
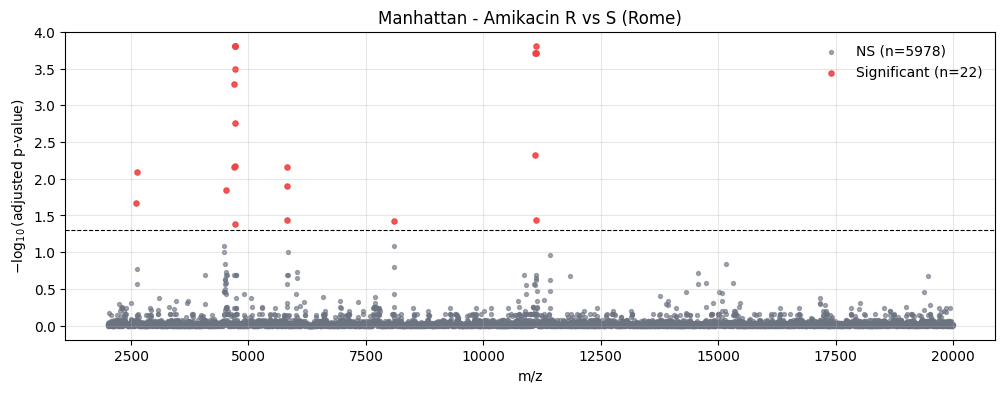
Manhattan Plot along the m/z Axis#
plot_manhattan places each bin at its m/z position and plots -log10(adjusted p-value). Points above the dashed line pass the significance threshold.
[7]:
from maldiamrkit.differential import plot_manhattan
_ = plot_manhattan(
analysis.results,
p_threshold=0.05,
title="Manhattan - Amikacin R vs S (Rome)",
)
Compare across Multiple Drugs#
DifferentialAnalysis.compare_drugs returns a boolean matrix whose rows are the union of significant m/z bins across all drugs and whose columns are drug names. True means the peak is significant for that drug.
We use the two antimicrobials annotated in MALDI-Kleb-AI - Amikacin and Meropenem - which share isolates but have different resistance mechanisms, so the overlap between their significant-peak sets is interesting on its own merits.
[8]:
ds_mero = load_maldi_kleb_ai(antibiotic="Meropenem", verbose=False)
encoder_nan = LabelEncoder(intermediate="nan")
y_mero = (
pd.Series(
encoder_nan.fit_transform(ds_mero.meta["Meropenem"]),
index=ds_mero.meta.index,
name="Meropenem",
)
.dropna()
.astype(int)
)
X_mero = ds_mero.X.loc[y_mero.index]
analysis_amk = DifferentialAnalysis(X, y).run()
analysis_mer = DifferentialAnalysis(X_mero, y_mero).run()
comparison = DifferentialAnalysis.compare_drugs(
{"Amikacin": analysis_amk, "Meropenem": analysis_mer},
fc_threshold=1.0,
p_threshold=0.05,
)
print(f"Union of significant bins: {len(comparison)}")
print("Per-drug counts:")
print(comparison.sum())
Union of significant bins: 14
Per-drug counts:
Amikacin 8
Meropenem 13
dtype: int64
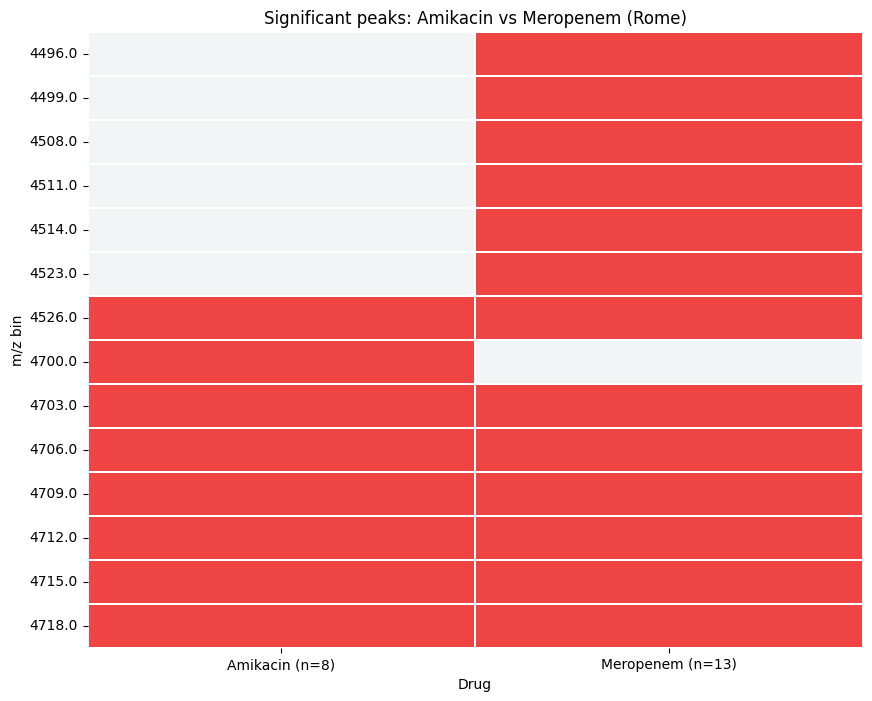
Boolean Heatmap#
plot_drug_comparison(..., kind='heatmap') renders a compact peaks x drugs matrix - useful for spotting drug-specific peaks at a glance.
[9]:
from maldiamrkit.differential import plot_drug_comparison
_ = plot_drug_comparison(
comparison,
kind="heatmap",
title="Significant peaks: Amikacin vs Meropenem (Rome)",
)
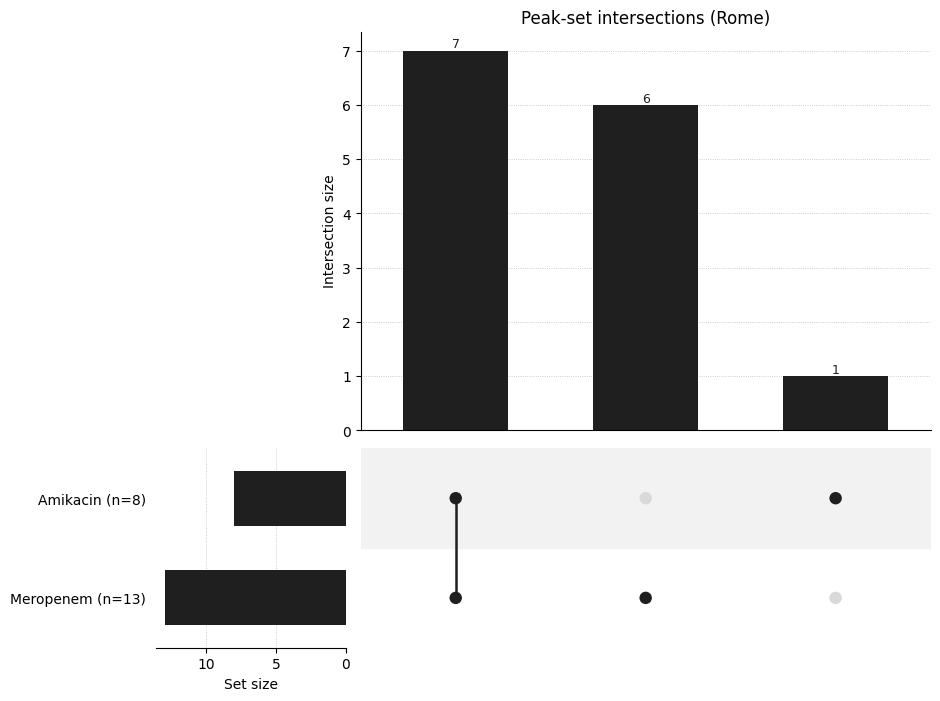
UpSet-style Intersection Plot#
With more drugs, a Venn diagram stops scaling - an UpSet plot shows the size of every intersection of significant-peak sets.
[10]:
_ = plot_drug_comparison(
comparison,
kind="upset",
title="Peak-set intersections (Rome)",
)
Choosing a Test and Correction#
DifferentialAnalysis.run() accepts both plain strings and the StatisticalTest / CorrectionMethod.
StatisticalTest enums:
|
When to use |
|---|---|
|
Default. Non-parametric, robust to MALDI intensity distributions. |
|
Welch’s t-test (unequal variances). Use for approximately-Gaussian data. |
CorrectionMethod enums:
|
Meaning |
|---|---|
|
Benjamini-Hochberg FDR. Good default. |
|
Benjamini-Yekutieli FDR. More conservative under dependence. |
|
Family-wise error control. Most conservative. |
Example of a conservative configuration:
[11]:
analysis_strict = DifferentialAnalysis(X, y).run(
test="t_test",
correction="bonferroni",
)
analysis_strict.top_peaks(n=5)
[11]:
| mz_bin | mean_r | mean_s | fold_change | p_value | adjusted_p_value | effect_size | |
|---|---|---|---|---|---|---|---|
| 3038 | 11114.0 | 0.000125 | 0.000080 | 0.652812 | 2.290201e-11 | 1.374120e-07 | 0.589519 |
| 905 | 4715.0 | 0.000506 | 0.000162 | 1.642962 | 9.656653e-11 | 5.793992e-07 | 0.546662 |
| 3039 | 11117.0 | 0.000127 | 0.000083 | 0.604224 | 2.661767e-10 | 1.597060e-06 | 0.558619 |
| 901 | 4703.0 | 0.000284 | 0.000120 | 1.241332 | 4.082946e-10 | 2.449768e-06 | 0.522023 |
| 3037 | 11111.0 | 0.000102 | 0.000067 | 0.604420 | 4.436762e-10 | 2.662057e-06 | 0.545698 |
Optional: Scoping Filters#
When you already know which m/z regions matter, or you want to limit the hypothesis burden, pass mz_ranges and/or peak_detector to run():
[12]:
from maldiamrkit.detection import MaldiPeakDetector
scoped = DifferentialAnalysis(X, y).run(
mz_ranges=[(3000, 6000), (9000, 12000)],
peak_detector=MaldiPeakDetector(method="local", binary=True, prominence=1e-4),
)
print(f"Bins tested (scoped): {len(scoped.results)}")
print(f"Bins tested (full run): {len(analysis.results)}")
print(f"#adj <= 0.05 (scoped): {(scoped.results['adjusted_p_value'] <= 0.05).sum()}")
Bins tested (scoped): 1994
Bins tested (full run): 6000
#adj <= 0.05 (scoped): 23
See Also#
Exploration notebook for PCA, t-SNE, UMAP, and spectral similarity